Анализ зарубежных и отечественных публикаций, посвященных биологическим и медицинским аспектам алкогольной зависимости, с очевидностью показывает, что проблема его происхождения и развития в течение последних десятилетий попала в тот замкнутый круг, который лишает практическую наркологию ее поступательного развития. Об этом говорит обилие патогенетических концепций и отсутствие, как рациональных мер профилактики, так и эффективной терапии алкогольной зависимости.
Этанол — естественный эндогенный метаболит, небольшие количества его всегда присутствуют в организме человека. В норме концентрация этанола в крови составляет 0,001-0,01%. Однократный прием 50-60 г чистого этанола, вызывающий состояние легкого опьянения и чувство эйфории, повышает концентрацию этанола в крови примерно, в 20 раз (до средних значений 0,2%). При концентрации этанола в крови 0,4% возможно наступление коматозного состояния, а уровень 0,7% приводит к остановке дыхания из-за паралича дыхательного центра.
Этанол быстро адсорбируется в желудочно-кишечном тракте; менее 1% его удаляется из организма через почки и легкие, а основная масса подвергается окислению в следующих реакциях. Алкоголь дегидрогенизируется, превращаясь в ацетальдегид [уксусный альдегид (АцА)], который под влиянием НАД и ферментов окисляется в уксусную кислоту. Остаток её в виде ацетил-КоА поступаетв цикл Кребса и окисляется в нём до СО2 и Н2О:
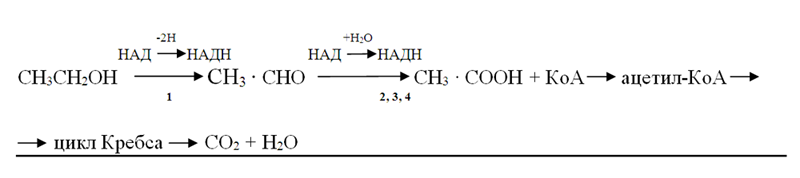
1 – алкогольдегидрогеназа (к.ф.1.1.1.1.); 2 – альдегиддегидрогеназа (к.ф. 1.2.1.3.); 3 – альдегидоксидаза (к.ф.1.2.3.1.); 4 – ксантиноксидаза (к.ф.1.2.3.2.).
Доминирующий путь окисления этанола (до 99% спирта) — приведенная выше алкогольдегидрогеназная реакция, которая протекает главным образом в цитоплазме клеток печени (Lieber C.S., 1988, 1994). Установлено, что фермент АДГ кодируется тремя генами. В тканях человека и животных он присутствует в виде димеров αα, ββ или γγ. Обнаружено около 20 изоформ этой дегидрогеназы. Их разнообразие обусловлено разными сочетаниями аллелей одного гена.
Установлено, что у людей разных рас спектр изоформ АДГ заметно отличается: для европейцев (кавказоидов) наиболее типичны изоформы АДГ2-2 — 1 (β1β1) и АДГ3-2 (γ2γ2), в то время как у лиц монголоидной расы (ориенталов) преобладают более активные изоформы АДГ2-1 (β2β1) и АДГ2-2 (β2β2). Иначе говоря, большинство монголоидов обладает АДГ с более высокой активностью, что обеспечивает быстрое окисление этанола и накопление АцА. У 50% лиц монголоидной расы ß-субъединица альдегиддегидрогеназы имеет передающуюся по наследству аномалию (аутосомно-доминантный тип наследования), значительно снижающую активность фермента. Поэтому у таких людей окисление АцА происходит очень медленно, концентрация этого метаболита в крови высока, что способствует развитию непереносимости к алкоголю (Ашмарин И.П. с соавт., 1999).
Очень небольшая часть этанола подвергается в пероксисомах печени действию каталазы (Ещенко Н.Д., 2004). Однако, по данным других исследователей (Абдукаева Н.С. с соавт., 2002) в пероксисомах печени под действием каталазы окисляется до 50% принятого внутрь этанола.
В окислении этанола принимает участие изофермент цитохрома Р450 (CYP2E1) — неспецифическая монооксигеназа. Реакция окисления протекает в микросомах (Щербаков В.М., Тихонов А.В., 1995).
В норме монооксигеназа окисляет менее 5% этанола. Однако при поступлении больших количеств алкоголя фермент индуцируется, и окислению в этой реакции подвергается до 70% этанола. Следует подчеркнуть, что резкое повышение активности монооксигеназы при алкоголизме приводит к значительному ускорению окисления ксенобиотиков, в том числе многих лекарственных препаратов. Этим объясняется повышенная чувствительность больных алкоголизмом к действию различных ядов. Кроме того, индукция монооксигеназы лежит в основе развития толерантности (устойчивости) к этанолу при его длительном потреблении.
Для того чтобы четко представить себе, как формируется алкоголизм, необходимо напомнить, что в организме человека и животных эндогенный этанол образуется из эндогенного ацетальдегида (АцА). Это означает, что в клетке с постоянством синтезируется и присутствует АцА, содержание которого колеблется от 0,19x10-9 до 0,27х10-9М. АцА в столь низких концентрациях, тем не менее, выполняет исключительно важную функцию регулятора тканевого дыхания (Бурбенская И.М., Ротенберг Ю.С., 1984), а его исчезновение или снижение его концентрации приводит к резкому падению потребления кислорода(Бурбенская И.М., Ротенберг Ю.С., 1986). В упрощенном виде эффект снижения содержания АцА (в плане потребления кислорода) напоминает эффект от введения несмертельной дозы цианида. Из-за особых химических свойств АцА, его способности даже без участия ферментов вступать в реакции с активными группами белков, липидов, сахаров, витаминов и др. он, в отличие от этанола, не может свободно перемещаться не только от клетки к клетке, но и даже от одних субклеточных структур к другим. Следует подчеркнуть, что если в какой-либо клетке прекращается синтез ацетальдегида, то соседняя клетка не может восполнить возникший его дефицит. В то же время хорошо известно, что АцА является высокотоксичным соединением, но поскольку он очень нужен клетке, в организме существуют мощные и строго организованные ферментные ансамбли, которые «следят» за поддержанием соответствующих низких концентраций этого соединения. Увеличение (в пределах нормы реакции) образования АцА сопровождается активацией дыхания, усилением процессов образования энергии и анаболических реакций и повышением функций любых клеток. При физиологическом снижении его концентрации активность клетки уменьшается. Поступление в организм алкоголя (этанола) извне, в первую очередь, приводит к изменению содержания в клетке АцА, поскольку у этанола нет иных путей превращения (Комисарова И.А., 1994).
С нарастанием дозы алкоголя (до 1,5-2 г/кг) и увеличением внутриклеточной концентрации АцА в 3-6 раз у него отчетливо обнаруживается свойство ингибироватъ важнейший ферментный комплекс, деятельность которого обеспечивает нормальное функционирование процессов дыхания и образования АТФ (Комисарова И.А. с соавт., 1986).
При дальнейшем увеличении концентрации у АцА проявляются свойства высокоактивного химического соединения и способность неспецифически взаимодействовать (связываться) с NH2-, SH- и СООН-группами белков с образованием различных стабильных и нестабильных соединений. Специфическое ингибирование большинства ферментов и является причиной появления наркотической фазы опьянения. В подобной ситуации самим же АцА «тормозятся» и ферменты, осуществляющие его детоксикацию. Присутствие АцА в тканях через 10-12 часов после опьянения формирует картину утреннего похмелья.
В связи с тем, что все клетки организма обладают альдегидокисляющими ферментами, при хроническом воздействии этанола их активность существенно увеличивается. Максимальная синхронизация и активизация альдегидокисляющих систем является основой формирования повышенной толерантности. Весьма важно отметить, что при хронической алкоголизации и при стабильно повышенной активности альдегидокисляющих ферментов достаточно сократить или прекратить потребление алкоголя – и соответственно уменьшить приток экзогенного АцА, как появляются симптомы его дефицита. Как уже указывалось выше, снижение концентрации АцА сопровождается резким сокращением потребления кислорода, нарушением окислительно-восстановительных реакций, процессов образования энергии и, соответственно, функций клеток всех тканей (мозга, миокарда, печени, почек, мышц). На фоне недостаточности АцА нарушение клеточных функций (Комисарова И.А. с соавт., 1984) и проявляется в форме алкогольного абстинентного синдрома (ААС). Именно недостаточностью АцА и можно объяснить идеальное купирование ААС этанолом и паральдегидом (триммером АцА). Следует обратить внимание на тот факт, что при алкоголизме на фоне депривации недостаточность эндогенного АцА может формироваться вследствие двух различных причин. В одном случае это обусловлено снижением его синтеза вследствие периодического накопления этого соединения за счет потребления алкоголя. Подобный вариант аналогичен явлению снижения образования, в частности, кортикостероидных гормонов или тироксина при лечении этими препаратами. Другой вариант недостаточности АцА (наиболее коварный) возникает при даже повышенном его эндогенном синтезе, но как следствие его быстрого окисления в уксусную кислоту за счет существенного увеличения активности альдегидокисляющих ферментов. Коварство такого рода относительной недостаточности заключается в том, что повышенный синтез эндогенного АцА как бы не позволяет «успокоиться» альдегидокисляющим ферментам, в результате чего в клетке не достигаются необходимые нормативные уровни концентрации АцА.
Каковы бы ни были молекулярные механизмы, приводящие к появлению недостаточности эндогенного АцА, но именно падение уровня его концентрации ниже нормы выполняет сущностную роль в формировании влечения к алкоголю.
Достижение физиологического баланса между синтезом АцА и его трансформацией или в эндогенный этанол или в уксусную кислоту, нормализующего уровень эндогенного АцА, в свою очередь, обусловливает появление ремиссии в динамике болезни.
Достаточно балансу между синтезом эндогенного ацетальдегида и его окислением нарушится, как возникнет рецидив заболевания. В зависимости от того, как долго организм способен «обеспечивать» этот баланс, будет находиться длительность ремиссии. И, наконец, если в клетке имеются возможности для быстрого восстановления физиологических концентраций АцА в случае падения его содержания, то возникающее влечение может купироваться организмом без применения алкоголя или лекарственных препаратов.
Следует указать, что ведущее значение в трансформации АцА принадлежит альдегидокисляющим ферментам, обладающим широкой субстратной специфичностью. Резко увеличивая свою активность при хроническом воздействии этанола, именно альдегидокисляющие ферменты никак не могут нормализовать свою активность и приблизиться к состоянию устойчивого биологического равновесия. В подобной ситуации деятельность этих ферментов (в силу широкой субстратной специфичности) может внезапно и существенно активизироваться под влиянием не только этанола и АцА, но и любых других альдегидов и соединений, образующихся в процессе обмена веществ. В первую очередь на альдегид окисляющие ферменты воздействуют биогенные амины, стероидные гормоны, которые, окисляясь, превращаются в альдегидные формы.
В соответствии с этим любые нагрузки (стрессовые, физические и т.п.), сопровождающиеся «выбросом» катехоламинов, стероидных гормонов, могут провоцировать при алкоголизме нарушение обмена АцА и рецидив заболевания (Комисарова И.А., 1994).
Среди множества эффектов этанола особенно важно действие его на нервную систему, поскольку именно нарушение функций ЦНС лежит в основе и формирования влечения к алкоголю и развития алкоголизма.
Масса клинических и экспериментальных данных свидетельствует о значительных морфологических и функциональных изменениях, происходящих в мозге при систематическом потреблении алкоголя: уменьшение массы мозга, истончение коры больших полушарий, увеличение расстояния между бороздами, расширение желудочков мозга (более выраженное у пожилых больных). В ряде исследований было отмечено также уменьшение массы маммилярных тел. Их разрушение предполагает ослабление кратковременной памяти и других когнитивных процессов, что часто наблюдается у больных алкоголизмом (Семке В.Я. с соавт., 2002).
Нейропсихологические исследования подтверждают характерное снижение когнитивных функций у этих больных, которое проявляется в дефиците абстрактного мышления, трудностях при решении зрительно-пространственных задач, нарушениях концептуальных оценок, расстройствах памяти. Тяжелым формам алкоголизма сопутствуют явные психоорганические нарушения в форме амнестических расстройств и алкогольной деменции. Для амнестических расстройств, связанных, по-видимому, с изменениями преимущественно в подкорковых структурах мозга, характерны нарушения оперативной и кратковременной памяти, некоторые изменения в поведении без нарушения сознания и общего снижения интеллекта (Пятницкая И.Н., 1994).
При алкогольной деменции резко снижаются интеллектуальные способности, нарушается память, абстрактное мышление и другие высшие функции мозга, что обусловлено нарушениями в кортикальных структурах (Фрэнсис Р.Д., 2000).
Нейропсихологические отклонения на ранних стадиях алкоголизма, проявляющиеся в виде изменений характера речи и функции активного внимания, снижении отдельных видов памяти, не являются необратимыми и могут быть восстановлены в период ремиссии или при отказе от алкоголя. Это подтверждается данными ЭЭГ.
Характерными симптомами алкоголизма являются тревожно-депрессивные состояния, агрессивность поведения (Волчегорский И.А., Мельник В.И., 2004). Установлено, что не только у человека, но и у животных этанол в значительно большей степени, чем другие вещества, редуцирует тревожно-депрессивные реакции.
В связи с этим особого внимания заслуживает анализ современных представлений о нейрохимической подоплёке тревожно-депрессивных расстройств (ТДР). В настоящее время большое внимание уделяется гипоталамической, гиперфункции кортикотропин-рилизинг-гормона (КРГ) в патогенезе депрессии major (Kathol R. et al., 1990; Slamecki C.J. et al., 1999; Solomon D.A. et al., 2000). Известно, что КРГ является центральным "медиатором" тревоги, агрипнии и анорексии. Важно подчеркнуть, что данный гормон обладает симпатомиметической, амфетаминоподобной активностью, которая в значительной степени определяет спектр его психотропных эффектов (Friedman E.M., Irvin M.R., 1995). Следует добавить, что "ложный медиатор" - ß-фенилэтиламин, тоже обладающий центральным симпатомиметическим действием, также способен вызывать тревожные расстройства поведения (Лапин И.П., 1988; Montegazza P., Riva M., 1963). Изложенное выше свидетельствует о целесообразности рассмотрения нейромедиаторных процессов в качестве основного, фармакологически управляемого, уровня развития ТДР.
Чем же ещё определяется такое пагубное воздействие алкоголя на нервную систему? Известно, что этанол обладает выраженной органотропностью: он накапливается в мозге, где концентрация его выше, чем в крови. Это связано с тем, что он легко проникает через ГЭБ и хорошо растворяется в липидах, которыми очень богаты ткани мозга (Шабанов П.Д., Калишевич С.Ю., 1998).
Из-за высокой растворимости этанола в липидах при повышении его концентрации в мозге происходят существенные изменения физико-химических показателей, характеризующих состояние мембранных структур. Меняется микровязкость и текучесть мембран, в фосфолипидном бислое появляется «свободное» пространство. Высокие концентрации этанола тормозят включение арахидоновой кислоты в фосфолипиды, но усиливают включение ее в триацилглицеролы, меняя таким образом качественный состав липидной компоненты мембранных структур. Этанол способен также взаимодействовать с длинноцепочечными жирными кислотами липидов с образованием этиловых эфиров, которые оказывают более сильный дезорганизующий эффект на мембраны, чем сам этанол. Подобные биохимические изменения сказываются на основных функциях мембранных структур нервной ткани, прежде всего таких, как генерация электрических потенциалов, ионные токи через мембраны и работа ионных каналов, высвобождение и обратный захват нейромедиаторов и др. (Мастеропуло А.П., Комисарова И.А., 1988).
Чувствительность мембранных структур мозга к действию этанола в определенной степени зависит от соотношения про- и антиоксидантных систем. Следует подчеркнуть, что высокая чувствительность мозга к повреждающему действию этанола коррелирует с большей интенсивностью процессов ПОЛ и меньшей активностью ферментов антиоксидантной защиты у крыс, предпочитающих алкоголь, чем у животных, предпочитающих воду (Lieber C.S., 1994).
Нарушение функций специфических мембран нейронов под действием этанола доказано нейрофизиологическими и нейрохимическими экспериментами, в которых зарегистрировано изменение мембранных токов Na+, К+, Са2+. В частности, проводимость потенциалзависимых Са2+-каналов по-разному меняется в зависимости от длительности алкоголизации. Однократное введение этанола снижает их проводимость и уменьшает вызванный деполяризацией вход Са2+ в нейроны. При хронической алкоголизации по мере развития толерантности к этанолу происходит постепенное повышение проводимости Са2+-каналов (Ross D.H., 1977; Meltzer H.L., 1986). При абстинентном синдроме проводимость потенциалзависимых каналов резко возрастает, усиливаются потоки ионов кальция в нейроны, что приводит к развитию возбуждения, столь характерного для абстинентного состояния. Эти экспериментальные наблюдения позволили не только обосновать, но и применить в клинике антагонисты потенциалзависимых Са2+-каналов для снятия повышенной возбудимости у больных алкоголизмом в состоянии абстиненции (Сидоренко Б.А., Преображенский Д.В., 1992; Del Pozo E. еt al., 1987).
При рассмотрении вопроса о влиянии этанола на метаболизм мозга следует обратить внимание на способность его резко тормозить всасывание витамина В1 (Сиволап Ю.П., 2006). Тиамин служит кофактором ряда декарбоксилаз, в том числе декарбоксилазного компонента пируват- и кетоглутаратдегидрогеназных комплексов. Так как пируватдегидрогеназа является одним из ферментов, лимитирующих скорость энергетического обмена мозга, вполне естественно, что при алкоголизме наступает резкое ухудшение энергетического статуса, приводящее к развитию алкогольной энцефалопатии.
Очень важным для понимания механизмов действия этанола на ЦНС является исследование влияния его на нейромедиаторные системы. К настоящему времени наиболее детально изучены эффекты алкоголя на катехоламин-, серотонин- и ГАМК-ергическую системы. На основании анализа изменений в этих системах под влиянием этанола выдвинута нейромедиаторная гипотеза формирования алкогольной зависимости (Herz A., 1998; Davidson D. et al., 1999; Grahame N.J. et al., 2000; Hall F.S. et al., 2001).
Показано, что даже однократный прием алкоголя повышает выброс норадреналина и дофамина в синаптическую щель, одновременно тормозя процесс обратного захвата медиаторов и снижая активность пресинаптических α2-адренорецепторов. Кроме того, замедляется метаболизм катехоламинов из-за снижения активности катехол-о-метилтрансферазы. Создающаяся при этом повышенная концентрация катехоламинов в синаптической щели вызывает ощущение эйфории, легкости, снимает эмоциональное напряжение, т. е. выступает как фактор внутреннего вознаграждения (Бондаренко Н.А. с соавт., 1981; Мосолов С.Н., 1995; Мюррей Дж., 1997).
Установлено также, что гиперактивность центральной и периферической норадренергической системы коррелирует с высоким уровнем личностной тревожности (Орликов А.Б., 1990). При этом фармакологические средства, угнетающие симпато-адреналовую систему (резерпин, клонидин, пропранолол), смягчают проявления тревоги (Крылов С.С., Старых Т.Т., 1973; Комисарова Р.А., Комиссаров И.В., 1990; Михайлов И.Б., 1999).
Накопление катехоламинов приводит через некоторое время к торможению их синтеза из-за ингибирования (по принципу обратной связи) тирозин- и дофамин-β-гидроксилаз, а также к ускорению окисления из-за повышения активности МАО. Наступающий в этих условиях некоторый дефицит норадреналина создает ощущение дискомфорта и побуждает к повторному приему алкоголя.
Иными словами, состояние физической зависимости от этанола в значительной степени определяется длительным ослаблением норадреналинергической системы из-за повышения активности ферментов, разрушающих катехоламины (МАО и КОМТ), увеличения обратного захвата медиатора и снижения чувствительности адренорецепторов (Анохина И.П., 1992).
При алкогольной зависимости из-за глубоких нарушений механизма обратной связи прекращение приема алкоголя приводит к избыточному выбросу катехоламинов. При этом особенно резко меняется уровень дофамина: прямые определения в СМЖ и крови показали более чем 10-кратное возрастание его концентрации (это связывают с развитием абстинентного синдрома). Ряд наблюдений над лабораторными животными позволил предположить, что в основе феномена предпочтения этанола могут лежать генетически детерминированные изменения в дофаминергической системе (Билибин Д.П., Дворников В.Е., 1991).
Таким образом, роль норадренергической системы проявляется в снятии эмоционального напряжения, формировании положительных эмоций и состояния эйфории при однократном приеме этанола, а также в формировании алкогольной мотивации на начальных этапах развития алкоголизма. В то же время, дофаминергическая система играет более выраженную роль при абстинентном синдроме, а нарушения ее могут лежать в генезе алкоголизма (Анохина И.П., 1992).
Следует подчеркнуть, что нормализация катехоламинергической нейромедиации в ЦНС также играет немаловажную роль в коррекции депрессивных расстройств. Так, например, ребоксетин (селективный ингибитор обратного захвата норадреналина) вызывает не только стойкую нормализацию настроения у больных с депрессивными расстройствами, но и обладает анальгетическим действием (Аведисова А.С. с соавт., 2003; Schüler P., 2002). Бромокриптин - специфический центральный агонист дофаминовых рецепторов типа Д-2 и прамипексол (высокоселективный стимулятор рецепторов Д-2, Д-3, Д-4 подтипов семейства Д-2 дофаминовых рецепторов) также снижают выраженность депрессии (Гехт А.Б. 2001; Горьков В.А., 2001; Обухова А.В., 2001).
Изменения активности основных ферментов метаболизма биогенных аминов под влиянием этанола могут влиять и на обмен серотонина. Известно, что серотонинергическая система рассматривается как один из компонентов системы внутреннего вознаграждения, и повышение уровня данного медиатора в определенных структурах вызывает положительные эмоции, ощущение благодушия и покоя. Вполне вероятно, что усиленная секреция серотонина даже после однократного приема алкоголя вносит свой вклад в ощущение эйфории и снятие эмоционального напряжения.
В то же время при синдроме абстиненции установлено резкое повышение активности серотонинергической трансмиссии, что может быть причиной галлюцинаций, характерных для этого состояния (Maycux R. et al., 1984; Fuller R.W., 1995).
В отдельных структурах мозга больных алкоголизмом установлено даже снижение концентрации серотонина. В этой связи следует отметить противоречивость сведений о роли серотонинергической системы. Подавление влечения к алкоголю зафиксировано для веществ, стимулирующих синтез серотонина или выброс его в синаптическую щель, а также для блокаторов его обратного захвата и агонистов (Вовин Р.Я. с соавт., 1992; Волошин В.М., 2003; Dimmork R.W. et al., 2000).
С другой стороны, в литературе имеются данные о подавлении потребления этанола и веществами, ингибирующими синтез серотонина или селективными антагонистами.
Такая неоднозначность данных говорит о том, что участие серотонинергической трансмиссии в эффектах этанола определяется в значительной мере типом (или подтипом) серотониновых рецепторов, а также их локализацией и вовлеченностью в патологический процесс на разных стадиях развития алкоголизма.
Из всех известных нейромедиаторных систем наибольшее внимание уделяется роли дисфункции ГАМК-ергических механизмов, как общей патогенетической основы ТДР (Смулевич А.Б. с соавт., 1997). Известно, что дефицит ГАМК-ергического торможения, сопряжённый с повышением возбудимости и избыточной импульсацией нейронов в диэнцефальных отделах мозга, закономерно вызывает состояние напряжённого беспокойства, т.е. тревоги (Калинин В.В., Максимова М.А., Недува А.А. с соав., 1996; Bursch G.P., Frings A., 1988).
В первую очередь дефицитарность ГАМК-ергической нейротрансмиссии связана с функциональной толерантностью ГАМК (А)-бензодиазепин-рецепторного комплекса (Мельник В.И., 2003; Волчегорский И.А., Мельник В.И., 2004).
Немаловажную роль в развитии этого феномена играют эндогенные анксиогены, являющиеся инверсными агонистами бензодиазепиновых рецепторов. К числу этих веществ относятся: широкая группа ß-карболинов и компонентов "трибулина" - изатин, этиловый эфир 4-гидроксифенилуксусной кислоты, 4-гидроксифенилэтанол и т.д. (Медведев А.Е. с соавт., 1995; Гловер В. С соавт., 1997; Волчегорский И.А. с соавт., 1997, 2000; Glover V. et al., 1982 Sandler M., 1982; Abel E.L., 1995). Вещества, обладающие трибулиновой активностью, способны вытеснять бензодиазепины из комплекса с их рецепторами. Известно также, что трибулинурия у больных, страдающих алкогольной аддикцией, превышает соответствующие показатели у здоровых людей (Sandler M., 1982).
Отдельного внимания заслуживает анализ анксиогенной роли продуктов кинуренинового метаболизма триптофана (Лапин И.П., 1970, 1976, 1980; Жуков В.Н. с соавт., 1985; Мельник В.И. с соавт., 1999). По данным ряда исследователей возбуждающие кинуренины (L-кинуренин и хинолиновую кислоту) целесообразно рассматривать не только в качестве потенциальных антагонистов бензодиазепиновых рецепторов, но также и блокаторов рецепторов ГАМК(А)-подтипа (Лапин И.П., 1983, 1997, 1998; Лапин И.П., Рыжов И.В., 1985; Рыжов И.В., Лапин И.П., 1986; Ерышев О.Ф., Рыбакова Т.Г., 1996; Petersen E.N. et al., 1983). Не удивительно, что бензодиазепиновые анксиолитики эффективно купируют развитие тревожных расстройств поведения, индуцированного у экспериментальных животных интерцеребровентрикулярным введением кинуренина (Лапин И.П., 1983; Орликов А.Б., 1990). В последние годы появились данные о том, что рецепторы ГАМК(Б)-подтипа могут также рассматриваться в качестве мишеней анксиогенного действия возбуждающих кинуренинов. При этом экранирование данных рецепторов специфическими агонистами (фенибутом и баклофеном) в большей степени предупреждает кинуренин-индуцированную тревогу, чем бензодиазепиновые анксиолитики (Мельник В.И., 1987; Мельник В.И., Макарчук В.А., Вострикова Э.П. с соавт., 1989; Мельник В.И., 1993; Мельник В.И., Вострикова Э.П., 1999; Волчегорский И.А., Мельник В.И., 2003; Волчегорский И.А., Власов А.А., Видякин В.А., Макарчук В.А., Мельник В.И. с соавт., 2003; Lapin I.P., 1996).
Следует подчеркнуть, что дефицитарность ГАМК-ергической нейротрансмиссии в последнее время рассматривается не только как нейрохимическая подоплёка тревоги, но и депрессии (Соловьёва В.М., Лонгинова С.В., 1986; Emrich H.M. et al., 1984). Установлено, что использование оптимальных дозировок бензодиазепиновых транквилизаторов способно оказать клинически значимый тимоаналептический эффект (Александровский Ю.А., 2003).
Большой интерес исследователей вызывает изучение влияния этанола на ГАМК-ергическую систему, в частности на работу сложного ГАМК(А)-рецепторного комплекса. На основании результатов целого ряда нейрофизиологических и биохимических экспериментов установлено, что этанол усиливает ток ионов хлора через мембранный канал ГАМК(А)-рецептора, т. е. усиливает ГАМК-ергическую передачу. Предполагается, что действие этанола связано с вызванным им изменением липидно-белкового окружения комплекса ГАМК — рецептор — хлорный канал (Алиев Н.А., 1989). Антагонисты ГАМК – пикротоксин и бикукуллин снимают ряд поведенческих эффектов этанола (седативное действие на поведение и координацию движений) (Аксентьев С.Б., Левинский М.В., 1990).
Ряд исследований с использованием обратимых агонистов [например, флумазенила (Ro15-1788)] позволил показать, что влияние этанола на работу хлорного канала ГАМК-рецепторного комплекса по крайней мере частично реализуется в результате действия его на бензодиазепиновый участок. Этанол повышает сродство данного участка к диазепаму и эндогенным эндозепинам. Эти результаты дополняют представления о механизмах седативного и анксиолитического (успокаивающее, снимающее тревожность) действия небольших доз этанола (Шабанов П.Д., 2002).
Для больных алкоголизмом характерно ослабление когнитивных способностей и памяти. Многие исследователи связывают это с действием этанола на глутаматергическую медиаторную систему, в первую очередь на рецепторы NMDA-типа (Masood K. et al., 1994). Как известно, именно они участвуют в механизмах формирования следов памяти, в процессах длительной постсинаптической потенциации. В опытах in vitro установлено, что алкоголь тормозит медиаторное действие глутамата на NMDA-рецепторы, причем степень замедления ионных токов (Na+, К+, Са2+) зависит от его дозы (Сиволап Ю.П., 2006). Установлено, что при действии ряда спиртов (метанол, этанол, бутанол, изопентанол), ослабление ионных токов, опосредованных NMDA-рецептором, пропорционально способности этих спиртов вызывать интоксикацию. К сожалению, конкретные детали действия этанола на NMDA-рецептор до сих пор не выяснены. Предполагается, что этанол блокирует ко-активирующее действие глицина, связываясь с регуляторным (глициновым) участком рецепторного комплекса (Шабанов П.Д., 2002).
Несмотря на достаточную противоречивость, весьма существенной и важной представляется роль пептидергических медиаторных систем в развитии алкоголизма. Установлено участие не только кортиколиберина, но и таких нейропептидов, как ангиотензин II, тиролиберин, брадикинин, в регуляции влечения к алкоголю. Показано, что под действием алкоголя изменяется ряд поведенческих эффектов пептидов. Кортиколиберин стимулирует развитие стрессорных реакций, а этанол в низких дозах снижает эффект кортиколиберина в экспериментах на моделях конфликтного поведения крыс. В модельных опытах при исследовании двигательной активности животных обнаружено, что тиролиберин уменьшает седативный и депрессантный эффекты алкоголя (Ещенко Н.Д., 2004).
Наиболее внятные сведения касаются взаимодействия этанола и опиоидергических систем, которые представляют собой важное звено механизмов внутреннего вознаграждения. Именно этот аспект давно привлекает внимание исследователей, пытающихся выяснить механизмы развития тяги к алкоголю (Blum K., Trachtenberg M.C., 1988).
Накоплен обширный массив данных о роли нарушении опиоидергических механизмов в развитии ангедонических расстройств при депрессии. Важно добавить, что героиновая аддикция, как и пристрастие к этанолу, зачастую формируется на преморбидно-депрессивной почве и приводит к усугублению депрессивных расстройств по мере формирования физической зависимости к опиоидам (Савченков В.А., Сиволап Ю.П., 2002; Мельник В.И., 2003).
В мозге животных, генетически предрасположенных к потреблению алкоголя, обнаружено меньшее содержание метэнкефалина и β-эндорфина, чем в норме; сниженный уровень β-эндорфина в гипоталамусе зарегистрирован и у животных после длительной алкоголизации. У людей даже однократный прием этанола приводит к повышению концентрации метэнкефалина в крови и ликворе, однако данные об изменении содержания β-эндорфина противоречивы. Для многих больных алкоголизмом характерен повышенный уровень антител к морфиноподобным соединениям в крови. Подобные наблюдения позволили предположить, что сниженный уровень эндогенных опиоидов в мозге обусловливает влечение к алкоголю как фактору, стимулирующему системы внутреннего вознаграждения (Blum K., Trachtenberg M.C., 1988).
Одним из доказательств участия опиоидной системы в формировании алкогольной зависимости служит то, что классический блокатор опиоидных рецепторов — налоксон оказывает положительный эффект при лечении не только алкоголизма и алкогольного абстинентного синдрома, но и острых алкогольных психозов (Буйков В.А., Мельник В.И. с соавт., 2002; Мельник В.И., Власов А.А. с соавт., 2002, 2003).
Весьма интересны результаты исследований непосредственного действия этанола на опиоидные рецепторы. Известно, что опиоиды могут модулировать острые и хронические эффекты этанола, а сам этанол способен модулировать эффекты опиоидов. При попытках разобраться в этом вопросе установлено, что in vitro этанол в высоких концентрациях (200-400 мМ), нарушая структуру липидной фазы мембран, прямо влияет на связывание опиоидов с рецепторами. Однако in vivo после приема этанола концентрация его в мозге в 2-4 раза ниже, поэтому возможность прямого модулирующего действия этанола на опиоидные рецепторы остается проблематичной, хотя установлено некоторое уменьшение аффинности δ-рецепторов и увеличение количества μ-рецепторов в мозге при сильном опьянении. В опытах с длительной алкоголизацией мышей найдено почти двукратное возрастание плотности δ-рецепторов.
Этанол способен не только вызывать изменения плотности и аффинности опиоидных рецепторов, но и влиять на сопряженные с ними системы внутриклеточной трансдукции сигналов. Так, на гомогенатах мозга и клеточных культурах установлено, что этанол повышает чувствительность аденилатциклазы к ингибирующему действию опиата эторфина. Обнаружено также изменение соотношения субъединиц G-белков в результате подавления этанолом экспрессии δ-субъединицы Gs-белка.
Действие этанола распространяется и на процесс образования и секреции эндогенных опиоидов. Об изменениях концентрации метэнкефалина и β-эндорфина в структурах мозга, СМЖ и крови при введении этанола было упомянуто ранее. Следует отметить, что влияние этанола на скорость синтеза, посттрансляционного процессинга белков-предшественников эндогенных опиоидов, а также на скорость секреции опиоидных пептидов не одинаковы в разных зонах мозга. Этим отчасти объясняется противоречивость данных литературы об изменениях концентрации того или иного опиоидного пептида в мозге под действием алкоголя.
И всё-таки, большинство авторов склоняется к точке зрения, что длительная алкоголизация животных, как и алкоголизм у человека, приводят к выраженным и стойким изменениям синтеза и секреции эндогенных опиоидных пептидов. Как правило, в этих условиях наблюдается снижение концентрации опиоидных пептидов в различных структурах мозга (Буров Ю.В., Ведерникова Н.Н., 1985).
Подтверждением роли опиоидной системы в действии этанола на мозг служит также возможность образования в организме эндогенных морфиноподобных соединений, которая резко возрастает при алкоголизме. Синтез морфиноподобных соединений происходит и при конденсации АцА с дофамином (сальсолинол) и при взаимодействии его с серотонином (метилтетрагидpo-β-карболин). Доказано, что сальсолинол и производные β-карболина в микроскопических количествах присутствуют и в нормальном мозге. Накопление АцА при алкоголизме резко усиливает эти реакции (Шабанов П.Д., 1999).
АцА тормозит окислительное дезаминирование дофамина, что способствует накоплению промежуточного метаболита – 3,4-диоксифени-лацетальдегида, который после конденсации с дофамином может через ряд промежуточных этапов превращаться в еще одно морфиноподобное соединение — норморфин. Продукты конденсации АцА с серотонином и дофамином обнаружены в мозге алкоголизированных животных, а также в крови и моче больных алкоголизмом.
Образующиеся с участием АцА эндогенные морфиноподобные соединения в зависисмости от их концентрации и ряда других условий действуют как агонисты или как блокаторы опиоидных рецепторов. Речь идет в первую очередь о субпопуляциях µ-рецепторов.
Продукты конденсации АцА и нейромедиаторов дофамина или серотонина, связываясь с опиоидными рецепторами, конкурируют с естественными их лигандами — эндогенными опиоидными пептидами, подменяя тем самым эндогенные факторы вознаграждения.
По мере развития алкоголизма и накопления сальсолинола и морфиноподобных соединений происходит изменение функционального состояния опиоидной системы: блокирование рецепторов в отношении эндогенных, наиболее адекватных факторов вознаграждения, появление субпопуляций модифицированных µ1- и µ2-рецепторов на фоне общего снижения чувствительности опиоидных рецепторов и возможного замедления образования эндогенных опиоидных пептидов. Это вызывает постоянное ощущение дискомфорта, неудовлетворенности и побуждает к дальнейшему приему алкоголя.
Говоря о роли пептидергических систем, нельзя не упомянуть о том, что действие некоторых нейропептидов на влечение к алкоголю в определенной мере коррелирует с участием их в развитии или ослаблении стрессорных состояний. Давно отмечено, что состояние стресса само по себе стимулирует потребление алкоголя. Мощный антистрессорный агент пептид дельта-сна (DSIP) при систематическом введении снижает потребление этанола экспериментальными животными, а инъекции этого пептида в желудочки мозга предотвращают развитие алкогольной мотивации у животных. Уровень DSIP достоверно ниже в стриатуме и плазме крови крыс, генетически предрасположенных к потреблению алкоголя, чем у животных, отвергающих этанол.
Подобные экспериментальные наблюдения указывают на то, что исходная недостаточность пептидных систем мозга, предохраняющих организм от стресса, может способствовать усилению тяги к алкоголю (Арушунян Э.Б., 2001).
Таким образом, накопленные к настоящему времени многочисленные данные, при всей противоречивости, позволяют всё-таки в общих чертах раскрыть сложные механизмы формирования алкогольной зависимости. Алкогольную мотивацию и формирование зависимости исследователи связывают с тревожно-депрессивными реакциями, гипофункцией ряда медиаторных систем (норадренергической, серотонинергической, ГАМК-ергической, опиоидной), изменением активности основных ферментов метаболизма этанола (АДГ и АльдДГ) и возможным положительным подкреплением этих систем при приеме этанола. Небольшие дозы этанола стимулируют функции нейромедиаторных систем, в то время как хроническое потребление алкоголя истощает, искажает, десинхронизирует, снижает их чувствительность.
Завершая обсуждение нейрохимических расстройств, их связь с ТДР, следует подчеркнуть, что выраженность этих расстройств существенно усугубляется у больных алкоголизмом в период отмены этилового спирта, клиническим следствием которой является алкогольный абстинентный синдром.